Massively Distributed Parallel EAs
Initially cEAs were designed to run in massively parallel
computers. Nowadays this kind of EAs is still studied in the literature due
to theirs special characteristics. As the massively parallel computers are disappearing
at present, the majority of the research in these algorithms is focused on the
sequential case, because when we try to make parallel implementations of cEAs
we find two important difficulties, which are characteristics
of cEAs:
- In cEAs there are a large number
of sub-algorithms because they have typically one single string in
every sub-algorithm.
- These sub-algorithms are tightly
connected.
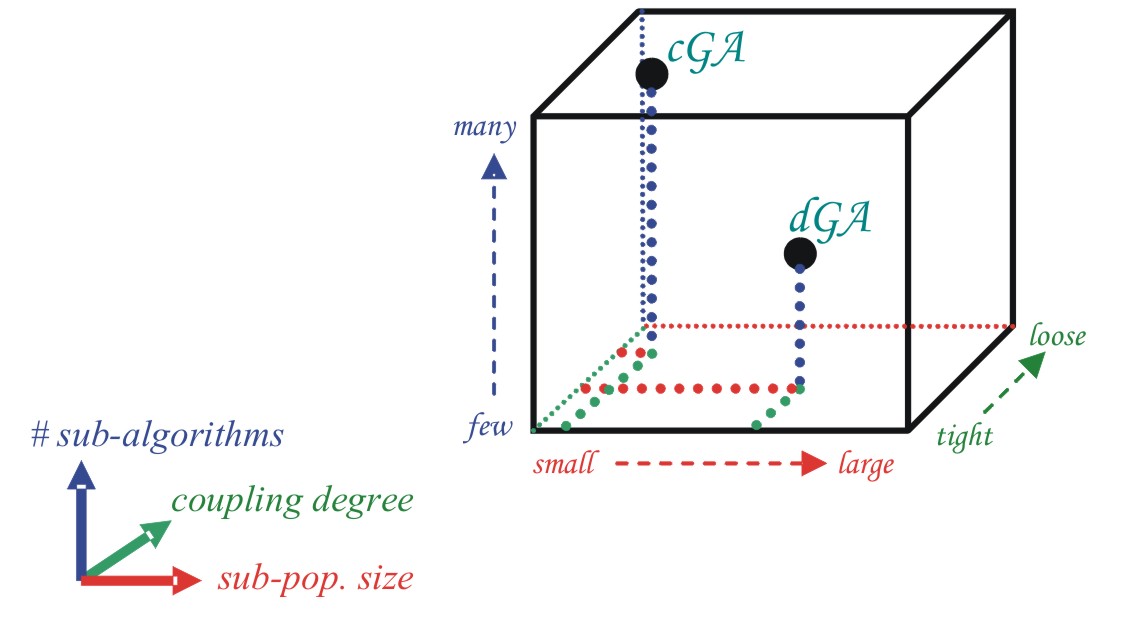
We can see in Figure 1 how cEAs have a very large number
of sub-algorithms, with small pop size each and a high coupling degree. In contrast,
we can see an easily parallelizable EA -dEA- with smaller number of sub-algorithms
running on bigger populations and with medium coupling degree.
|
| Figure 1. The structured-population genetic
algorithm cube |
However we can find in the literature many studies trying
to parallelize cEAs. The motivation for these works
are the possibility of facing problems which are limited
in CPU, getting better solutions in less time,
making profitable the available hardware resources,
etc. On the other hand, parallelizing cEAs may
be complex because they have a very high connectivity
among individuals.
Following we will expose six different
strategies for parallelizing fine-grained EAs. The first two approximations
are parallel fine-grained panmistic EAs (not decentralized), while the four
later correspond to parallel cEAs:
- This first algorithm was proposed by Maruyama, Hirose,
and Konagaya in [MHK93].
In this fine-grained algorithm each processor has only one active individual
and a buffer to store K suspended individuals. Search in this algorithm
progresses by repeating the cycle below (there are no synchronization).
- Each processor sends out a copy of its active individual to another
processor which is selected at random.
- For each of the individuals that have arrived from other processors.
- One of the suspended individuals in the buffer is deleted according
to their relative fitness if the number of the suspended individuals
is equal to K.
- The newly arrived individual is stored in the buffer.
- The recombination operator is applied to the active individual. A part
of the active individual is replaced by a part of one of the suspended
individuals which have not been used for the recombination yet.
- The mutation operator is applied to the active individual.
- The active individual is evaluated and compared with the suspended individuals
in the buffer.
- If the active individual can not survive the selection, one of the suspended
individuals is selected according to their relative
fitness and exchanged with the active individual.
- This micro grained model is the master and slave model
[HMT00]. In
this model, simple GA except the evaluation operation is performed in the
master processor and transfers the data to the slave processors. The master
transfers the data of one gene to one processor and the slave evaluate the
fitness of the gene and return the data to the master. Just after the master
receive the data from the slave, the master send the data of new gene. This
algorithm is summarized in Figure 2.
Forgaty et al.[FH91]
reported that this model is very effective when the number of population
size is huge. Maruyama et al. [MHK93]
developed the efficient program of this model. The advantage of this
model is that the program coding is very simple. Even when there are
huge number of genes, users need not to change the programs. The other
advantage is that it can be easy to keep the load balance.
On the other hand, the parallel efficiency does not become 100 %because
of the existence of the master CPU. Therefore, when there are not
so many processors, it is a very big disadvantage.
PGA
pack is a free software
of this model. |
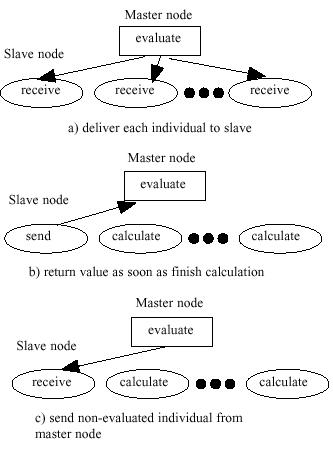 |
| Figure 2. The flow
of fine grained model |
|
- This third implementation uses a circularly linked linear
ordering of subpopulations [Balu93].
Each subpopulation evolvse only 2 chromosomes per generation. These 2 chromosomes
are chosen from a group of 10 chromosomes. The group is comprised of 1 chromosome
from each of the four immediate left and 1 chromosome from each of the four
immediate right subpopulations, and the 2 chromosomes which were evolved in
the subpopulation during the previous generation.
Each chromosome selected from the neighbors is chosen randomly from
the two evolved at each neighbor. The fitness of every chromosome is calculated
relative to the other chromosomes in the group of 10. Two chromosomes from
the set of 10 are probabilistically chosen for recombination, based upon
their relative fitness. The other 8 chromosomes are discarded.
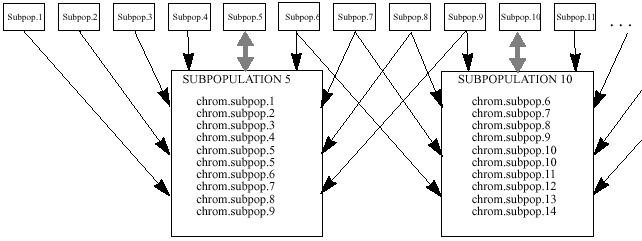 |
| Figure
3. Baluja's architecture of subpopulations, arrangement #1. Subpopulation
5 and 10 are shown enlarged. They randomly select 1 chromosome from the
2 chromosomes in each of their 8 closest neighbors, 4 from the left neighbors
4 neighbors, 4 from the right. They also incorporate their own two chromosomes
into the population. From this population two chromosomes are probabilistically
chosen for breeding. The 8 chromosomes not selected are discarded. |
- The fourth implementation uses a two dimensional toroidal
array of subpopulations [Balu93].
Each of the subpopulations evolves 2 chromosomes which are chosen from a group
of 10. The 8 immediate neighboring subpopulations donate to the group of 10.
The remainder of the procedure is completed in the same manner as described
above.
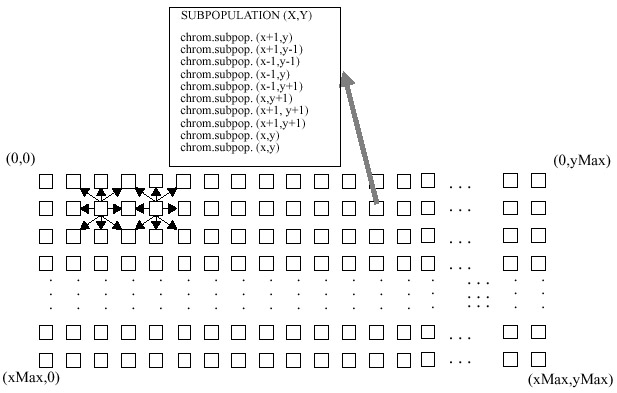 |
| Figure 4. Baluja's architecture of subpopulations,
arrangement #2. A two dimensional array of processors is assumed. Each processor
contributes one of its two chromosomes to each of its 8 nearest neighbors.
A sample population for processor (x,y) is shown. The processors are connected
in a toroid. |
- In the fifth implementation [Balu93],
a linear ordering of subpopulations is used once again. One chromosome from
each of the 3 immediate left and one chromosomes from the 4, 5, 6 subpopulations
from the right contribute to the group of 10 chromosomes. Unlike the first
implementation, the immediate right subpopulations are not used. Since there
are 4 remaining positions in the group of 10, they are filled with 2 copies
of each of the chromosomes evolved in the previous generation. After the selection
of 10 chromosomes, the algorithm proceeds in the same manner as described
earlier. This model allows a faster spread of chromosomes than the first implementation,
and slower rate than the second one.
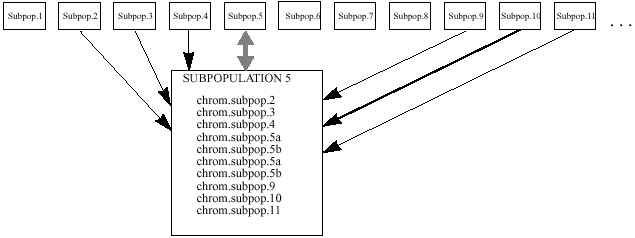 |
| Figure 5. Baluja's architecture of
subpopulations, arrangement #3. Subpopulation 5 is shown enlarged. |
- This last algorithm is an implementation of a
diffusion model GA. It was proposed by White and Pettey at [WP97].
The diffusion model GA typically maps each individual in the population to
a different process. Each process is then executed on its own processor. In
order to perform selection and crossover, each process must communicate with
other processes to gather information about other individuals. This communication
causes overhead that slows down the execution of the GA. Thus, selection and
crossover are usually performed with the knowledge of only a subset of the
population, often called the neighborhood.